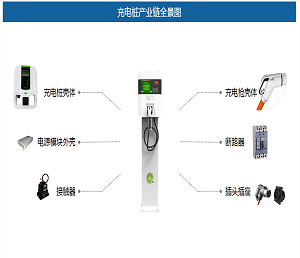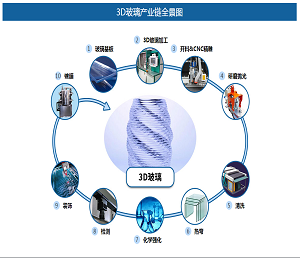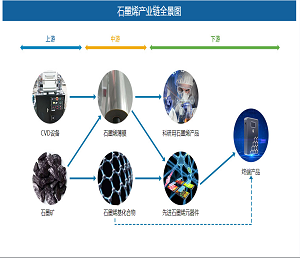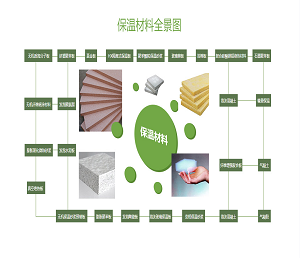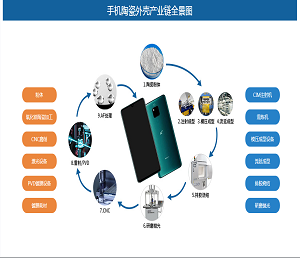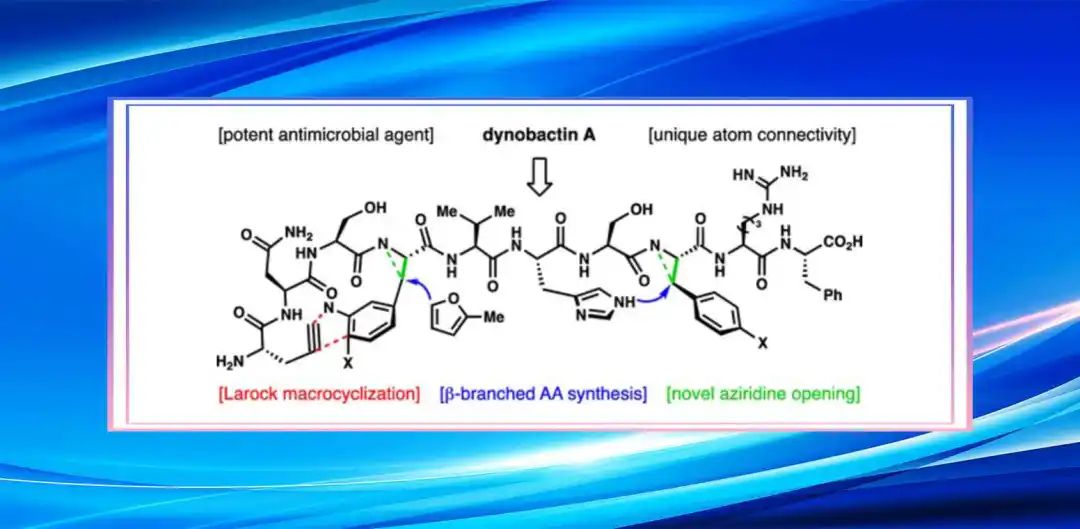

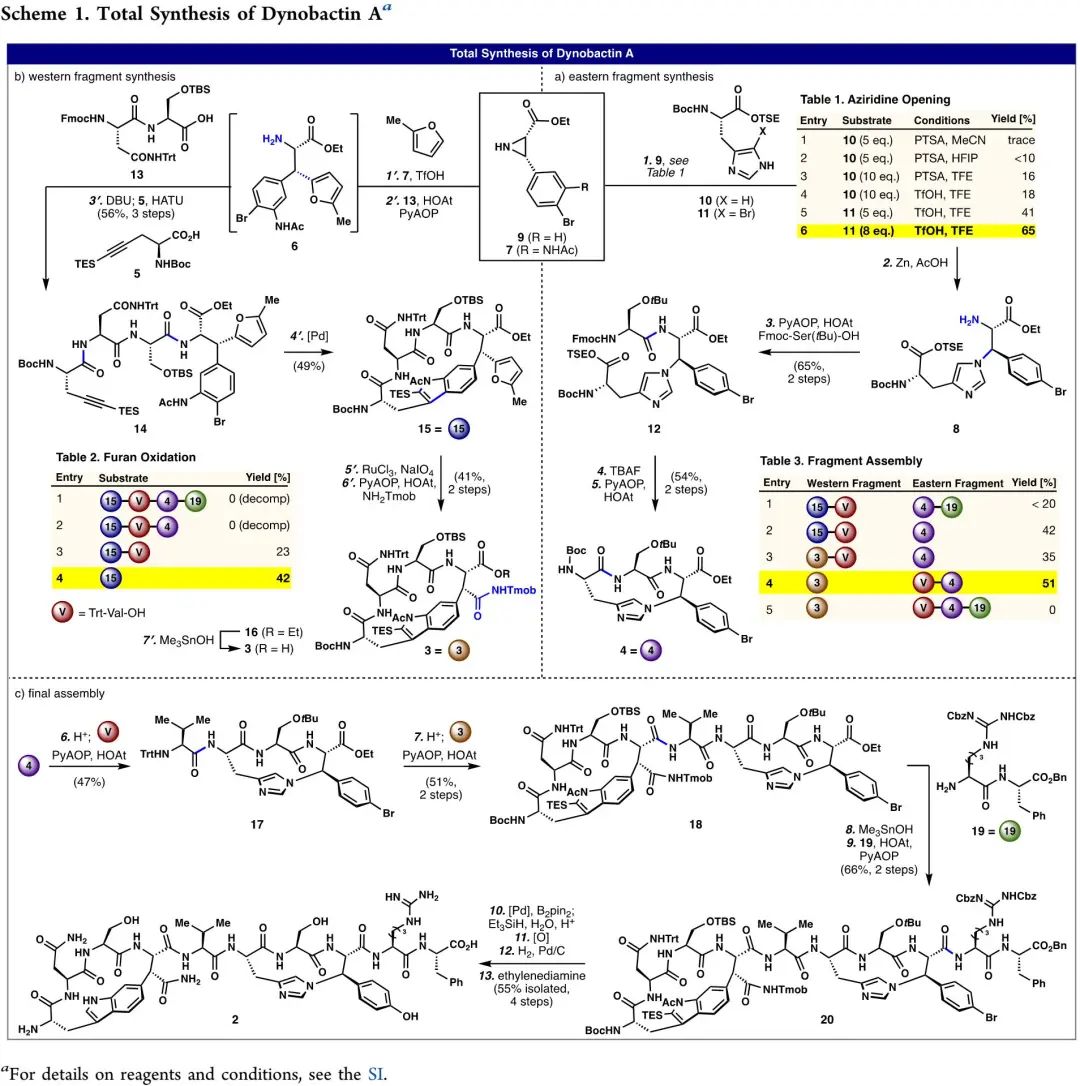
最近,美国斯克利普斯研究所Phil S. Baran课题组报道了强效抗生素dynobactin A的首次全合成(LLS 16步)。该研究工作以呋喃基和对溴苯基作为天然产物中最敏感一级酰胺和对苯酚替代物,利用氮杂环丙烷开环策略构建两个独立的β-芳基氨基酸片段,实现dynobactin A的汇聚式全合成。相关研究成果发表在近期的《美国化学会志》上(J. Am. Chem. Soc. DOI: 10.1021/jacs.3c11560)。
天然产物darobactin A(1)和dynobactin A(2)是最近发现的两个低细胞毒性抗生素(Figure 1),对革兰氏阴性菌表现出强效抗菌活性,能通过突变避开细菌耐药性。两个天然产物都以BamA蛋白为靶点,BamA蛋白是BAM复合物中的一种细菌插入酶,能促进革兰氏阴性菌外膜蛋白的折叠和插入。相比稠合双环七肽分子1,dynobactin A(2)是由两个大环通过肽键连接起来的非稠合双环十肽分子。这种非稠合双环结构赋予dynobactin A更大的分子柔性和额外的离子电离位点,使其水溶性是分子1的20倍以上。此外,2中的C21位、C39位立体中心构建难度极大。总之,dynobactin A(2)具有的复杂生物活性和独特分子结构,使其合成极具挑战性。
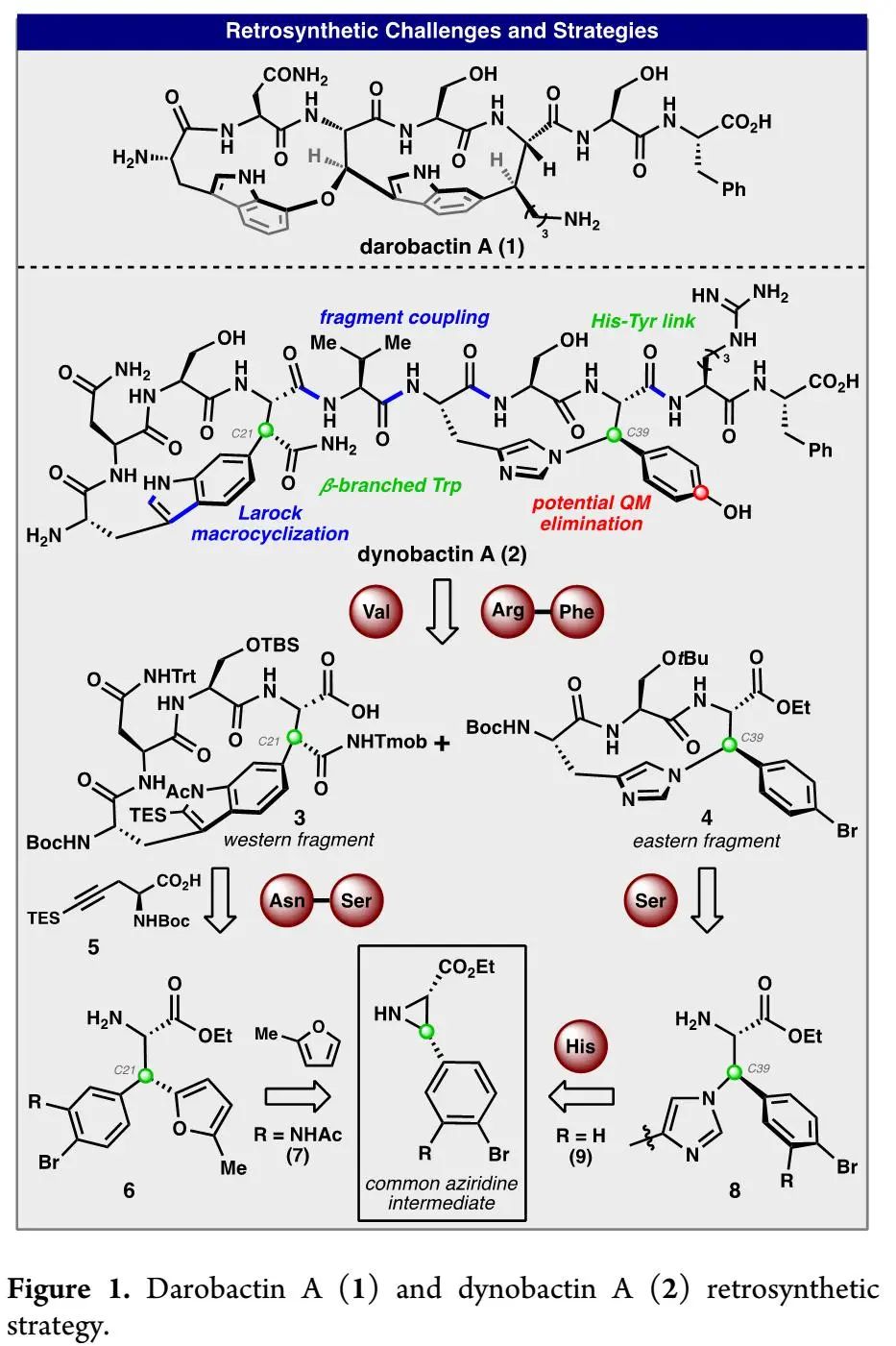
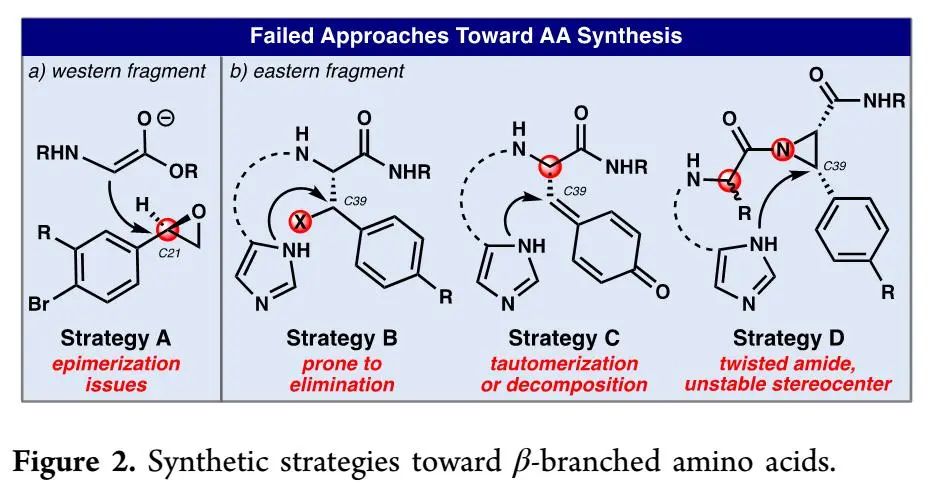
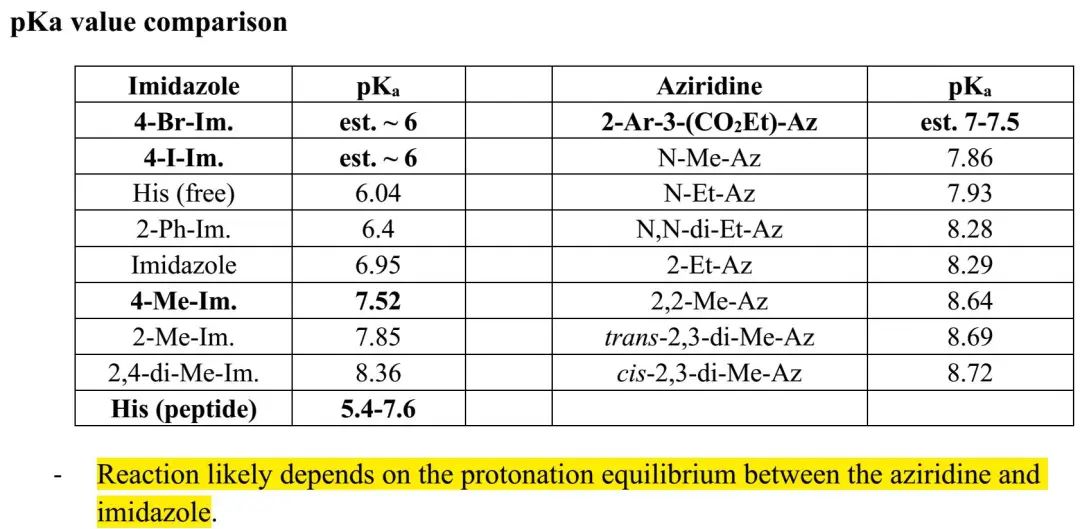
为了实现dynobactin A的全合成,首先要合成AA片段6和9(β-branched amino acid),为此作者开展了大量AA合成策略尝试。如Figure 2所示,西侧片段或东侧片段的A-D四种合成策略,都未能实现AA片段的理想合成。为了合成西侧片段,作者尝试了亲核性烯醇化物和环氧化物间的开环反应(A策略),但所得开环产物β-立体中心在后续反应中会发生异构化。为了合成东侧片段,作者尝试了三种策略但均不理想:1)咪唑片段和β-卤代酪氨酸发生亲核取代反应(B策略),易发生消除生成不饱和脱氢氨基酸或发生底物降解;2)咪唑片段和酪氨酸衍生的醌化合物发生仿生分子内环化反应(C策略),易发生底物异构化或降解;3)组氨酸上咪唑片段和N-酰基氮杂环丙烷发生开环反应(D策略),受酰基氮杂环丙烷的“扭曲酰胺”特征影响,易发生丝氨酸ɑ-位立体中心异构化或消除等副反应。
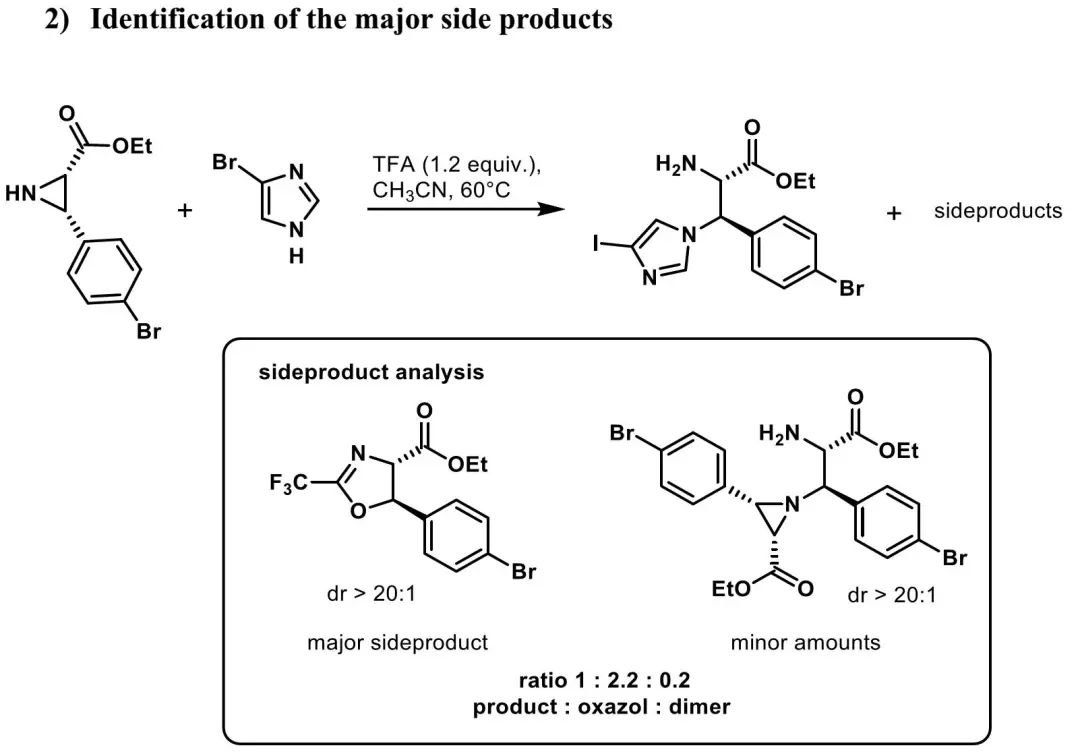
本文授权转载自微信公众号「CBG资讯」,未经许可谢绝二次转载,如需转载请联系C菌(微信号:chembeango101)